Introduction
This third article in a series on genotoxic/mutagenic impurities (GTIs) will discuss the challenges facing synthetic and analytical chemists as they endeavor to identify and ultimately control levels of these GTIs or PGIs (potentially genotoxic impurities) in drug substance and drug products. GTIs are residual reagents, intermediates, degradation products or by-products of synthesis that have a demonstrated ability to damage DNA, thus having the potential to cause a mutagenic and possibly a carcinogenic response [1]. GTIs are typically electrophilic (e.g. alkyl halides, alkyl sulfonates, epoxides [2]), or can be metabolized to an electrophilic moiety (e.g. aromatic amines, hydrazines [3]) and act predominantly by alkylation of DNA bases. PGIs are compounds containing an alerting structural motif; moieties such as hydrazine, epoxide, aziridine and N-nitrosamine confer a high likelihood of mutagenic activity, whereas others (e.g. N-oxide, carbamate, Michael acceptor, carboxylic acid chloride, methylol) are considered to be either false positives or extremely weak alerts [4].
Risk Management
It is never possible to truly avoid all risks associated with GTIs/PGIs and thus the conventional strategy of avoid > reduce (As Low as Reasonably Practicable, ALARP) > control (Threshold of Toxicological Concern, TTC) is in many ways untenable. This is because reactive reagents and intermediates act as key ‘building-blocks’ required to synthesize complex, multi-functional drug substances, and it is practically impossible to avoid using or creating GTIs/PGIs in a typical synthesis. It may be possible to re-design the synthesis to avoid a specified GTI/PGI, but the alternative synthesis will in all probability have other GTI(s)/PGI(s) that may also need to be eliminated or controlled.
A further consideration is the application of the TTC (Threshold of Toxicological Concern for cancer endpoints) default specification limit for a GTI/PGI of 1.5 μg/day. The use of this highly conservative metric, equivalent to a virtually safe dose (VSD) for lifetime exposure, is often unnecessary and/or inappropriate. This particular TTC was originally developed in relation to a threshold of regulation for potentially carcinogenic food ingredients/components (at 0.15 μg/day), but it is not possible to legislatively eliminate numerous naturally occurring genotoxic and carcinogenic substances in food. Bruce Ames [5] has endeavored to publicize the fact that the daily human exposure to carcinogenic natural pesticides (secondary plant metabolites formed in order to deter predation, numbering 5000-10000 different compounds) is up to 1.5g/day.
So although there are significant potential risks from dietary and other carcinogens, human exposure is often difficult or impossible to control. On the other hand, although GTIs/PGIs in drug substance/ drug products constitute a much lower risk, patient exposure can be controlled. Such control is allied with a pragmatic acceptance of some level of risk, currently defined as a likelihood of ≤ 1 in 100,000 of excess cancer risk over a patient’s entire lifetime [6]. This acceptance of risk is also enshrined within ICH Q9 [7], which articulates the necessity for quality risk management, as part of an overarching Quality by Design (QbD) strategy (ICH Q8 [8]) for both drug substance and drug product pharmaceutical processes.
The regulatory risk model as it applies to GTIs/PGIs has been challenged however. Bouder [9] argues that this is not the most favored approach and that a “Tolerability of Risk” (ToR) strategy is more suitable, where ‘tolerable’ does not equate to ‘acceptable’, but rather indicates a ‘willingness by Society as a whole to live with a risk so as to secure certain benefits, in confidence that the risk is worth taking and that it is being properly controlled.’ That is, where an appropriate risk/benefit balance has been derived, and where overall the benefit to the patient outweighs the risk. This principle is incorporated to some extent (but without quantitative guidance on tolerable levels of GTIs) in ICH S9 [10], which applies to anticancer medicinal products. In oncology products used in advanced cancer indications, it is possible to make a case for managing GTIs/PGIs as ordinary impurities in accordance with ICH Q3A/ Q3B [11, 12]. However, a recent survey of pharmaceutical companies [13, 14], highlighted that 50% of participating companies still applied strict controls to GTIs in cytotoxic oncology products, thus demonstrating a level of ambiguity in interpretation of the guidance. In contrast, 82% of participating companies did control GTIs when developing non-cytoxic/ non-genotoxic oncology drugs.
The TTC [6, 15] and the related staged-TTC [15, 16] provide a range of working limits in terms of defining ‘acceptable risk’ versus the stage of clinical development (see Table 1) and in relation to duration of exposure for marketed products.
Table 1. Staged TT C for Medicinal Products (derived from [15, 16])
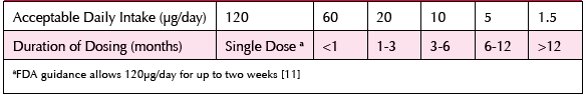
Hence, for risk management purposes, the key focus is to assess the likelihood of any GTIs/PGIs exceeding the TTC /staged TTC in a drug substance/drug product and where such a liability is identified as being theoretically possible, to provide adequate assurance of effective control strategies for those GTI(s)/PGI(s). A PGI, defined by EMA as “an impurity that shows a structural alert for genotoxicity, but that has not been tested in an experimental test model” may or may not be present in the drug substance [15]. Thus an impurity could be either potentially genotoxic or not, and/or the impurity could theoretically be present or absent in final API. The ‘worst case’ scenario applies when an impurity is both Amespositive (mutagenic) and actually detected in the drug substance.
Genotoxic Risk Assessment (GRA)
The optimal risk assessment process is a multi-displinary (chemistry/ toxicology), drawing on in-silico as well as in-cerebro (understanding and experience) prior-knowledge approaches. Computer-based expert systems,structure activity relationships (SARs) and expert knowledge on synthetic chemistry and genetic toxicology can all be used in a complementary fashion, with additional input, as required, from formulation, analytical and regulatory sciences. The overall initial objective is to evaluate the synthesis of the drug substance and assess those impurities that ‘might be reasonably likely to occur (i.e. potential impurities) for genotoxic potential’ [6]. A similar exercise can be applied to drug-product impurities based on data from stability studies and theoretical considerations. The decisions on which structures to submit for GTI assessment(s) appear to be both straightforward and reasonably consistent across the pharmaceutical industry [17].
The EMA [6] and FDA [15] draft guidelines recognize the utility within SAR assessment of in-built toxicophores as a valid process for assessment of genotoxic potential. The use of in-silico systems has gained a level of regulatory acceptability. An open-source system developed mainly for industrial chemicals, but also applicable to pharmaceutical impurities, is CAESAR (Computer Assisted Evaluation of industrial chemical Substances according to Regulations) [18]. “Standard” in-silico systems are designed to minimize false-negative predictions and so in some cases an impurity with a positive prediction may, on testing in an Ames assay, produce a negative result as mentioned in our earlier article [4] for the artificial sweetener, sucralose. The EMEA Q&A supplement [16] confirmed that no action is required for any unidentified impurity below the ICH Q3A [11]/Q3B [12] identification thresholds. Hence there is no requirement to identify and assess every unknown sub-threshold impurity observed within the API and the resulting formulated product. Any impurity that does not trigger an Ames alert based on visual inspection and/or an appropriate SAR system is dealt with according to the ICH Q3A/B guidelines for synthetic impurities. Impurities that do contain an alert (PGIs) can be handled using the staged TTC/TTC approach, if appropriate; however, it is strongly advisable to carefully evaluate any alerting impurity in other ways such as searching the literature for mutagenicity data on appropriate model compounds, or actually testing in an Ames assay, before proceeding on the assumption of a staged TTC/TTC limit. Most pharmaceutical companies utilize supplementary internal proprietary databases that may contain existing genotoxicity data on the compound as such or on structurally similar compounds [17]. Thus prior knowledge can overrule a structural alert, at least in theory, although regulators may in some cases require results from a confirmatory Ames test. Similarly, GTIs may also be human metabolites and the risk assessment needs to assess the implications of this scenario [6]. One PGI example is nicotine N-oxide, which is a recognized impurity in nicotine as well as being a human urinary metabolite [19], but which is Ames-negative [20].
However, ‘qualification’ of a GTI via this route (even if based on a ‘clean toxicology package for the parent’ [21]) can be problematic from the regulatory perspective if the metabolite is a confirmed mutagen.
As mentioned above, the GRA process starts with the assessment of the API synthetic route for postulated and/or known impurities using structural alerts as the key determinant, i.e. identification of PGIs. At this stage there are two key unknowns: Is the PGI actually present in the drug substance? And secondly, is the PGI actually genotoxic, i.e. Ames positive? An additional critical downstream consideration is whether carcinogenicity bioassay data are available on the PGI/GTI, or on a closely related compound. If so, a compound-specific risk assessment, as described previously [4], should be feasible without recourse to using the TTC limit. An Industry Taskforce (under the auspices of PhRMA) proposed that the PGIs would be classified into one of five different categories, dependent on a combination of experimental and comparative structural assessment [22]. These classes are summarized in Table 2 (where class 1 has the most significant cause for concern; whereas, class 5 has the least cause for concern).
Table 2: Division of PGIs into Different Classes Dependant on Experimental and Comparative Structural Assessment (based on Muller et al., 2006 [22])

Extreme caution should be exercised in using this and other similar schemes since the labels “genotoxic” and/or “carcinogenic” are often used indiscriminately and may not apply to the impurity in question. In the context of GTIs, “genotoxicity” has a specific meaning of a positive result in the Ames assay. For example, subsequent more thorough evaluations may negate earlier positive reports (e.g. ethyl bromoacetate), or a reaction product of the compound with the solvent used for Ames testing (most frequently DMSO) may be the cause of the positive result (as with thionyl chloride and many other sulfonic and carboxylic acid chlorides) [23]. Some non-genotoxic carcinogens have been mistakenly tagged as GTIs and the TTC limit applied, e.g. benzene, acetamide, etc.
 Figure 1: Listing of Structurally Alerting Functional Groups using Ashby-Tennant Criteria
Figure 1: Listing of Structurally Alerting Functional Groups using Ashby-Tennant CriteriaA range of “classical” alerting molecular fragments are shown in Figure 1, although as explained in the introduction, there is considerable doubt as to the applicability of some of these in light of experimental data obtained over the last two decades. Dobo et al. [24] indicated that the most common structurally alerting motifs in pharmaceutical impurities were aromatic nitro, aromatic amines, Michael acceptors and alkylating agents; and less commonly, epoxides, oximes and hydrazines.
The main focus of the GRA is on impurities that may arise particularly in the penultimate and final synthetic stages (generally excluding purification steps) and this should take into account interactions between reagents and solvents. Such reactions may often be deactivating such as the hydrolysis of any residual acid halide under aqueous conditions. Another much publicized example is the potential reaction between a short-chain alcoholic solvent such as methanol or ethanol and a sulfonic acid leading to formation of an alkyl methane sulfonate during sulfonic acid salt synthesis from the base form of the API [25]. In fact, recent publications have established that alkyl sulfonate formation will not occur under normal conditions using a molar equivalent amount of sulfonic acid since sulfonate ester formation is exceedingly slow and salt formation is essentially instantaneous, since the rate of the proton transfer reaction is diffusion-controlled [25, 26]. The GRA is then completed by a review of possible control strategies for the agreed GTIs/ PGIs and finally establishment of actual control measures where required. This process needs to be iterative and re-visited whenever changes (in synthetic route, in starting material supplier, etc) are being considered.
Chemical Purging Assessments
It is relatively easy to perform a GRA on a synthetic route and identify all of the theoretical PGIs (or GTIs) that could be present in the final API. Such an approach produces a significant number of theoretical PGIs and additional chemical reasoning can be used to evaluate their significance, particularly if they arise at an early stage in a long synthetic route [27]. This kind of exercise can be based on the known (or projected) reactivity of a particular GTI/PGI in conjunction with the known downstream chemistry (subsequent to the formation of the specified GTI/PGI). The chemical information is used to evaluate the likelihood that the GTI/PGI in question will be purged from the synthesis prior to its incorporation into the final API.
Unfortunately, the above type of chemical purging analysis is the subject of significant misunderstandings between Industry and the regulators. Regulatory agencies routinely perform paper-based GRA determinations of the synthetic route, but then may request proof of absence of all identified theoretical PGIs (or GTIs) from the API, and take no account of downstream chemistry capability of purging these PGIs.
The responses provided by QWP (Quality Working Party) to specific issues of interpretation of the CHMP guideline on genotoxic impurities raised by EFPIA (European Federation of Pharmaceutical Industries and Associations) highlighted the issue of purging of GTIs/PGIs as an area of misinterpretation [28]. In one of the case studies regulators asked: ‘are data available to confirm that PGIs are removed by chemical purging?’ This was a specific example where arguments based on chemical and scientific reasoning were not accepted as part of the initial regulatory assessment. QWP disagreed with the regulatory assessment and indicated in this case that the scientific rationale for adequate purging can be accepted, and that, more generally, supporting experimental data may not always be needed. In a second example, there were two impurities that raised SAR alerts (DEREK /MCASE), and it was argued, based on chemical and scientific reasoning, that both impurities would be effectively purged from the synthesis. However, the regulatory authority still requested compliance with CHMP quality guidelines. QWP again disagreed with this interpretation and indicated that the question should not have been raised and that the scientific rationale was acceptable.
Some industry experts have been keen to provide regulators with some structure for their GRA risk assessment of the synthetic processes. Teasdale’s et al. [29] semi-quantitative assessment process based on physicochemical parameters allows for a predictive purge factor to be defined. Based on this tool, the authors argued that they could predict, which, if any, GTIs/PGIs are likely to be present in the final API and then focus attention on the most likely impurities and identify which chemistries are likely to be effective in controlling relevant processes and resultant impurity levels. This approach is fully aligned with the tenets of risk assessment as defined in ICH Q9 [7].
Snodin [26] argued that highly reactive reagents (possessing a structural alert for genotoxicity) that are utilized in the early stages of complex synthetic pathways are unlikely to be carried over to the final API. Similarly, Pierson et al. [27] reasoned that most PGIs arising from reagents or synthetic intermediates which occur four, or more, synthetic stages upstream of the API are likely to be eliminated or significantly reduced by the downstream chemistries and processes. This perspective has received some support from the regulators. In the recent EMA Q&A update [16], the EMA does describe different scenarios based on where the impurity is introduced into the synthetic route. If the impurity is introduced before the final stage of the synthesis then it can be omitted from the final API specification if it can be demonstrated (using spiking experiments), that levels do not exceed 30% of the TTC, staged TTC, or otherwise defined acceptable limit. In contrast, if the impurity is introduced into the final stage of the synthesis then it should be included in the final API specification. Skip lot testing can be applied if the GTI does not exceed 30% of the proposed limit.
Alternatively, assessments can be performed experimentally (typically post-PoC), often facilitated by spiking of the specified GTI at elevated levels, to facilitate analytical determination and aid the tracking of the GTI through subsequent synthetic stages. This is often termed spiking/purging or impurity fate mapping. This approach was well illustrated by Liu et al. [30] for pazopanib hydrochloride. In order to implement an effective QbD strategy for the in-process control of five designated GTIs, comprehensive spiking/purging experiments were performed on the API synthesis [30]. This procedure demonstrated the maximal purging capacity of the process, and the data were used to define the in-process limits for all GTIs and to justify not performing testing on the final API (QbD versus Quality by Testing). To understand process tolerability, all five GTIs were spiked into stage 1 at varying levels, up to a maximum of 5% (equivalent to 50,000 ppm), and residual levels of each GTI were monitored at subsequent stages and also in the API. This approach is exemplified using one of the specified GTIs, i.e. Impurity II.
For Impurity II spiked at 5% in Stage 3, the final product yields a concentration less than that of the final product specification (1.7 ppm). The significant decrease is attributed to several processing steps that assist with consuming the impurity. This information allowed higher specification settings for the impurities investigated at earlier phases in the synthetic process, without the need for final product testing [30].
Based on these purging experiments in-process controls were implemented for all five GTIs and these in-process controls were set at percentage levels (rather than ppm levels), which in turn were aligned with HPLC/UV methodology. Therefore, this QbD approach ensured that five sophisticated HPLC/MS methods for testing residual levels of the five GTIs in the final API could be replaced by simplified HPLC/UV methods, which are much more aligned with quality control in a production setting; without any reduction in the overall quality of the final API [30].
Analytical Method Development
As a result of the need for PGI/GTI analyses at very low levels, i.e. typically in the ppm to sub-ppm range (driven by staged TTC/ TTC limits), it is necessary to have an appropriately sensitive analytical method to support early-stage development. Since developing such assays is a resource-intensive exercise, it is advisable to fully evaluate the mutagenicity risk before embarking on this activity. This is especially true for reactive intermediates and unstable degradation products, where analyte stability is an issue, and for drug products, where matrix effects constrain the method’s ability to achieve appropriate sensitivity. Another significant challenge is that these sensitive methods need to be developed rapidly for early phase development candidates (pre-Phase I) and this has prompted the development of ‘generic’ methods for common analytes, e.g. sulfonate esters [31]. Numerous articles have appeared in the scientific literature mainly based on the analysis and testing of GTIs (or PGIs) in APIs. However, these methods can typically be modified to address the problems of interference from the drug product matrix [32], particularly when supplemented by selective extraction techniques such as LLE [liquid liquid extraction; SPE (solid phase extraction); SPME (solid phase microextraction] or matrix deactivation.
In addition to the very low GTI (or PGI) levels within the sample and the associated issue of matrix interference (particularly problematic for drug product impurities, i.e. degradants), the key analytical challenge is often the divergent nature of the GTI (or PGI) alerting structural motifs which often preclude generic common analytical approaches. Examples from the literature are either focussed on summarizing the different analytical approaches applicable to specific alerting structural groups; e.g. sulfonate esters [33], alkyl halides [34], epoxides/ hydroperoxides [35], hydrazines [36], or generic methods applicable to any API/drug product/excipients within that specific structural class, e.g. sulfonate esters using headspace (HS) GC-MS [30], or HILIC (hydrophilic interaction liquid chromatography)-ESI (electrospray)- MS (mass spectrometry) [37], hydrazines by GC-MS [38], volatile aldehydes by GC-MS [39], alkyl chlorides by HS-GC-ECD (electron capture detector) [40]. Recently, Sun et al. [41] have published a systematic method development strategy for the determination of GTI (or PGI) contaminants in APIs. They advocate the use by first intent of sophisticated GC/MS (/MS) or HPLC/MS (/MS) methodologies. These methods can be simplified to GC-FID/ECD or HPLC/UV approaches post-PoC (Proof of Concept), when greater process understanding aligned with QbD initiatives allow for the GTIs to be controlled using in-process specifications to higher levels, aligned with ICH Q3A/ ICH Q3B (0.05% ≡ 500ppm), predicated on downstream chemistries purging the reactive GTIs from the final API [42].
Although technology transfer of these sophisticated, sensitive hyphenated methods into a manufacturing environment is possible [43], there are significant concerns about the long term reliability of such methods in production environments. In particular, the need for highly specialized equipment and supporting infrastructure, the need for equipment maintenance and the need for a high technical knowledge base. Some companies have made the strategic decision to keep such methods within R&D and accept the resulting greater regulatory scrutiny (pre approval inspections/cGMP inspections).
Conclusions
Dealing effectively with PGIs/GTIs calls for a high level of technical expertise particularly in chemistry and toxicology. Simplistic approaches, for example applying the TTC limit to any PGI, are likely to be counterproductive and generate a significant amount of unnecessary inputs in relation to process changes, analytical method development and API purification. The various techniques described in this short article are complementary in nature and serve firstly to narrow down the number of PGIs under consideration (based on purge factor analysis and API analysis), secondly to establish which key PGIs are actually mutagenic (in the Ames assay) based on public-domain or proprietary data, and thirdly to determine appropriate limits for specific GTIs using generic metrics such as the TTC or staged TTC, or by performing a compound-specific risk assessment (normally using carcinogenicity bioassay data).
Acknowledgements
Jim Harvey (GSK) for discussions around the genotoxicity of sulfonyl chlorides and carboxylic acid chlorides and Andy Teasdale (AZ) on the risk assessment tool.
References
- A.R. Lookyer, M.P. Ryan, B.J. Neubert-Langille, R. Naji, R. Risk Assessment of Potentially Genotoxic impurities within the Framework of Quality by Design, Organic Process R&D, 14; 1015-1020 (2010).
- E. Sobol, M.E. Engel, E. Rubitski, W.W. Ku, J. Aubrecht, R.H. Schiestl, Genotoxicity profiles of common alkyl halides and alkyl esters with alkylating activity, Mutation Research, 633; 80 (2007).
- T. Leakakos, R.C. Schank, R.C. Hydrazine toxicity in the neonatal rat, Toxicology Applied Pharmacology, 126; 295 (1994).
- D.Snodin and D.P.Elder. Genotoxic Impurities Part II: Toxicology Overview, Pharmaceutical Outsourcing, 13(5); 38-50 (2012).
- B.N. Ames, M. Profet, L.S. Gold, Nature’s Chemicals and Synthetic Chemicals: Comparative Toxicology, Proc. Natl. Acad. Sci. U.S.A., 87; 7782-7786 (1990).
- Guidelines on the Limits of Genotoxic Impurities, EMEA Committee for Medicinal Products for Human Use, CPMP/SWP/5199, London, England, 2006.
- ICH Q9. Quality Risk Management, In International Conference on Harmonization, Harmonized Tripartite Guideline; ICH Expert Working Group; Geneva, Switzerland, 2006.
- ICH Q8. Pharmaceutical DevelopmentIn International Conference on Harmonization, Harmonized Tripartite Guideline; ICH Expert Working Group; Geneva, Switzerland, 2009
- F. Bouder, Regulating Impurities in Pharmaceutical Products: A Tolerability of Risk Approach? Expert Rev.Clin. Pharmacol., 1; 241- 250 (2008).
- ICH S9. In International Conference on Harmonization, Harmonized Tripartite Guideline; ICH Expert Working Group; Geneva, Switzerland, 2006.
- ICH Q3A, Impurities in New Drug Substances, in International Conference on Harmonization Tripartite Guideline. Current Step 4 version 2.
- ICH Q3B, Impurities in New Drug Products, in International Conference on Harmonization Tripartite Guideline. Current Step 4 version 2.
- S.T. Colgan, G.Mohan, G.Vudathala, P.Kaspar, M.McMshon, US Regulatory Rapporteur, 7(5); 23-27 (2009).
- S.T. Colgan, G.Mohan, G.Vudathala, P.Kaspar, M.McMshon, US Regulatory Rapporteur, 7(6); 21-23 (2009).
- Guidance for Industry, Genotoxic and Carcinogenic Impurities in Drug Substances and Products: Recommended Approaches, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), December 2008
- European Medicines Agency, Questions and Answers on the ‘Guideline on the Limits of Genotoxic Impurities’, EMA/CHMP/ SWP/431994/2007 revision 3, 23rd September 2010 (accessed on 21st December 2010).
- K.L.Dobo. In silico methods combined with expert knowledge rule out mutagenic potential of pharmaceutical impurities: An industry survey, Regulatory Toxicology Pharmacology, 62; 449-455 (2012).
- Anon. CAESAR (Computer Assisted Evaluation of industrial chemical Substances according to Regulations). http://www. ncbi.nlm.nih.gov/pmc/articles/PMC2913329/. Accessed on 09th January 2013.
- N.L. Benowitz, J. Hukkanen, P. Jacob. Nicotine chemistry, metabolism, kinetics and biomarkers. 3rd.Handb. Exp. Pharmacol., 192; 29-60 (2009).
- D.J. Doolittle, R.Winegar, C.K. Lee, W.S.Caldwell, A.W.Hayes, J.D.de Bethizy. The genotoxic potential of nicotine and its major metabolites, Mutatation Research, 344(3-4); 95-102 (1995).
- E.Klenke. A regulatory perspective of genotoxic metabolites, Infoma Life Sciences conference on Genotoxic Impurities, 29th June 2012, Zurich, Switzerland.
- L. Mueller, R.J. Mauthe, C.M. Riley, M.M. Andino, D De Antonis, C. Beels, J. DeGeorge, A.K.M. De Knaep, D Ellison, J.A. Fagerland, R. Frank, B. Fritschel, S. Galloway, E. Harpur, C.D.N. Humfrey, A.S. Jacks, N. Jagota, J. MacKinnon, G. Mohan, D.K. Ness et al., A Rationale for Testing, and Controlling Specified Impurities in Pharmaceuticals that Posses the Potential for Genotoxicity, Reg. Tox. Pharm., 44; 198-211 (2006).
- S.Thea, G. Cevasco. A novel reaction of benzoyl chlorides in dimethyl sulfoxide, J. Org. Chem., 53; 4121-4122 (1988).
- K.L. Dobo, N. Greene, M.O. Cyr, S. Caron, W.W. Ku, The Application of Structure Based Assessment to Support Safety and Chemistry Diligence to Manage Genotoxic Impurities in Active Pharmaceutical Ingredients during Drug Development, Regul. Toxicol. Pharmacol., 44; 282-293 (2006).
- D.P.Elder, K.Facchine, J.N. Levy, R. Parsons, D. Ridge, L. Semo, A. Teasdale, An Approach to Control Strategies for Sulfonate Ester Formation in Pharmaceutical Manufacturing Based on Recent Scientific Understanding, Organic Process R&D, 16; 1707- 1710 (2012).
- D.J. Snodin, Genotoxic Impurities: From Structural Alerts to Qualification, Organic Process R&D, 14; 960-976 (2010).
- D.A. Pierson, B.A. Olsen, D.K. Robbins, K. DeVries, D.L. Varie, Approaches to Assessment, Testing Decisions, and the Analytical Determination of Genotoxic Impurities in Drug Substances, Organic Process R&D, 13; 285-291 (2009).
- European Medicines Agency, QWP Responses to EFPIA Paper: Industry Experiences of the Implementation of the EMA Guideline on the Limits of Genotoxic Impurities (Quality Related Aspects), EMA/INS/440301/2010, 07th July 2010, London.
- A.Teasdale, S.Fenner, A. Ray, A. Ford, A. Philips. A Tool for the Semiquantitative Assessment of Potentially Genotoxic Impurity (PGI) Carryover into API Using Physicochemical Parameters and Process Conditions, Organic Process R&D, 14; 943-945 (2010).
- D.Q. Liu, T.K. Chen, T.K, Analytical control of genotoxic impurities in the pazopanib hydrochloride manufacturing process. J. Pharm. Biomed. Anal., 50; 144-150 (2009).
- R. Alzaga, R.W. Ryan RW, K. Taylor-Worth, A.M. Lipczynski, R. Szucs R, P.A. Sandra. A generic approach for the determination of residues of alkylating agents in active pharmaceutical ingredients by in situ derivatization-headspace-gas chromatography-mass spectrometry. J. Pharm. Biomed. Anal., 45; 472-479 (2007).
- Sun M, Bai L, Terfloth GJ, Liu DQ, Kord AS. Matrix deactivation: A general approach to improve stability of unstable and reactive pharmaceutical genotoxic impurities for trace analysis. J. Pharm. Biomed. Anal., 52(1); 30-36 (2010).
- D.P. Elder, A. Teasdale , A.M. Lipczynski. Control and analysis of alkyl esters of alkyl and aryl sulfonic acids in novel active pharmaceutical ingredients. J. Pharm. Biomed. Anal., 46; 1-8 (2008).
- D.P. Elder, A. Teasdale ,A.M. Lipczynski. Control and analysis of alkyl and benzyl halides and other related reactive organohalides as potential genotoxic impurities in active pharmaceutical ingredients. J. Pharm. Biomed. Anal., 48; 497-507 (2009).
- D.P. Elder, D. Snodin D, A.Teasdale. Analytical approaches for the detection of epoxides and hydroperoxides in active pharmaceutical ingredients, drug products, and herbals. J. Pharm. Biomed. Anal., 51; 1015-1023 (2010).
- D.P. Elder, D. Snodin D, A.Teasdale. Control and Analysis of hydrazine, hydrazides and hydrazones – Genotoxic Impurities in Active Pharmaceutical Ingredients (API) and drug products, . J. Pharm. Biomed. Anal., 54; 900-910 (2011).
- J. An, M. Sun, L. Bai , T. Chen, D.Q. Liu DQ, A. Kord. A practical derivatization LC/MS approach for determination of trace level alkyl sulfonates and dialkyl sulfates genotoxic impurities in drug substances. J. Pharm. Biomed. Anal., 48; 1006-1010 (2010).
- M. Sun, D.Q. Bai L, Liu. A generic approach for the determination of trace hydrazine in drug substances using in situ derivatizationheadspace GC-MS. J. Pharm. Biomed. Anal., 49; 529-533 (2009).
- Z. Li Z, B.M. Kozlowski , E.P. Chang. Analysis of aldehydes in excipients used in liquid/semi-solid formulations by gas chromatography-negative chemical ionization mass spectrometry. J Chromatogr. A, 1160; 299-305 (2007).
- G.K. Ellinson G.K. Generic method for residual alkyl chlorides in active pharmaceutical ingredients. Pharmaceutical analysis science group (PASG) (Autumn meeting). 2-10-2006. Milton Keynes UK.
- M. Sun M, D.Q. Liu DQ, A. Kord. A Systematic Method Development Strategy for Determination of Pharmaceutical Genotoxic Impurities. Organic Process R&D, 14; 977-985 (2010).
- D.Q. Liu DQ, M. Sun M, A.S. Kord. Recent advances in trace analysis of pharmaceutical genotoxic impurities. J. Pharm. Biomed. Anal., 51; 999-1014 (2010).
- P.J. Borman, M.J. Chatfield, E.L. Crowley. Development, validation, and transfer into a factory environment of a liquid chromatography tandem mass spectrometry assay for the highly neurotoxic impurity FMTP (4-(4-fluorophenyl)-1-methyl-1,2,3,6-tetrahydropyridine) in paroxetine active pharmaceutical ingredient (API). J. Pharm. Biomed. Anal., 48; 1082-1089 (2008).
Dr. David Snodin studied Chemistry at Imperial College, London, moving to Bristol University to undertake research for a Ph.D. in Organic Chemistry. He later obtained an M.Sc. in Toxicology at the University of Surrey. He has been involved in regulatory toxicology for over 30 years, initially in industry, then at the UK Medicines and Healthcare products Regulatory Agency (Expert Preclinical Assessor and UK representative on the CHMP Safety Working Party). He was recently employed for 8 years by PAREXEL International Drug Development Consulting as Vice President – Nonclinical. In February 2010 he formed his own consulting business – Xiphora Biopharma Consulting, specializing in nonclinical consulting generally with a particular interest in the Quality-Safety interface (particularly involving safety assessment of impurities and excipients) and environmental risk assessment. His recent publications are focused on the safety evaluation of genotoxic and nongenotoxic pharmacopoeial and other impurities.
Dr. David Elder studied chemistry at Newcastle upon Tyne, before moving to Edinburgh University to undertake research for a Ph.D. in Crystallography. Dr. Elder has 34-years of experience in the pharmaceutical industry at a variety of different companies (Sterling, Syntex and GSK). For the last 19 years, he has been employed by GSK. He is currently a director in the externalization group (Scinovo) in GSK R&D. Dr Elder is a member of the British Pharmacapoeia (Expert Advisory Group PCY: Pharmacy), a member of the Analytical Division Council (Royal Society of Chemistry, UK) and a council member of the Joint Pharmaceutical Analysis Group, UK. He is a member of the PhRMA and EfPIA sub-groups on genotoxic impurities and was part of the PQRI group that assessed the control strategies for alkyl mesilates. His recent publications are focused on the chemistry/analytical evaluation of genotoxic impurities. He has authored three chapters in the recently published Genotoxic Impurities: Strategies for Identification and Control (Ed. Andrew Teasdale, Wiley, 2010).